Trích:
|
Nguyên văn bởi Dinh Tien Dung
Anh ơi cho em hỏi PM3 và AM1 là cái gì thế anh?
|
AM1
From Wikipedia, the free encyclopedia
• Find out more about navigating Wikipedia and finding information •
Jump to: navigation, search
AM1, or Austin Model 1, is a semi-empirical method for the quantum calculation of molecular electronic structure in computational chemistry. It is based on the Neglect of Differential Diatomic Overlap integral approximation. Specifically, it is a generalization of the modified neglect of differential diatomic overlap approximation. Related methods are PM3 and the older MINDO.
AM1 was developed by Michael Dewar and co-workers and published in 1985. AM1 is an attempt to improve the MNDO model by reducing the repulsion of atoms at close separation distances. The atomic core-atomic core terms in the MNDO equations were modified through the addition of off-center attractive and repulsive Gaussian functions.
The complexity of the parameterization problem increased in AM1 as the number of parameters per atom increased from 7 in MNDO to 13-16 per atom in AM1.
The results of AM1 calculations are sometimes used as the starting points for parameterizations of forcefields in molecular modelling.
AM1 is implemented in the MOPAC, AMPAC, GAUSSIAN, GAMESS (US) and GAMESS (UK) programs.
PM3 (chemistry)
From Wikipedia, the free encyclopedia
• Ten things you didn't know about Wikipedia •
Jump to: navigation, search
PM3, or Parameterized Model number 3, is a semi-empirical method for the quantum calculation of molecular electronic structure in computational chemistry. It is based on the Neglect of Differential Diatomic Overlap integral approximation.
The PM3 method uses the same formalism and equations as the AM1 method. The only differences are: 1) PM3 uses two Gaussian functions for the core repulsion function, instead of the variable number used by AM1 (which uses between one and four Gaussians per element); 2) the numerical values of the parameters are different. The other differences lie in the philosophy and methodology used during the parameterization: whereas AM1 takes some of the parameter values from spectroscopical measurements, PM3 treats them as optimizable values.
The method was developed by J. J. P. Stewart and first published in 1989. It is implemented in the MOPAC program (of which the older versions are public domain), along with the related RM1, AM1, MNDO and MINDO methods, and in several other programs such as GAUSSIAN, GAMESS (US), GAMESS (UK), Chem3D, Spartan, AMPAC, and BOSS.
The original PM3 publication included parameters for the following elements: H, C, N, O, F, Al, Si, P, S, Cl, Br, and I. Many other elements, mostly metals, have been parameterized in subsequent work.
A model for the PM3 calculation of lanthanide complexes, called Sparkle/PM3, was also introduced.
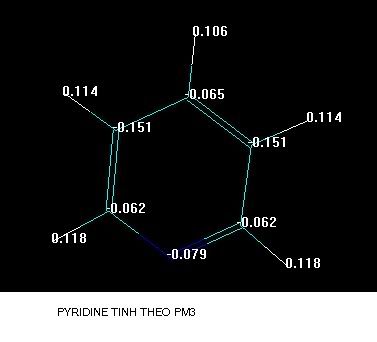
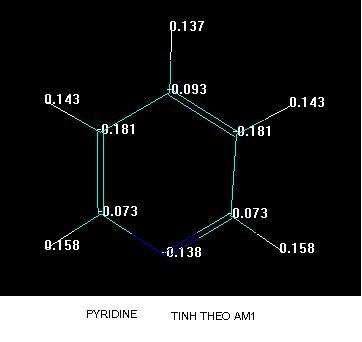
Đây là hai phương pháp tính toán bán kinh nghiệm mà hay dùng khi tối ưu cho các phân tử hợp chất Hữu cơ,












 Zero
Zero






.gif "Matcuoi (28)")
.gif "Matcuoi (9)")

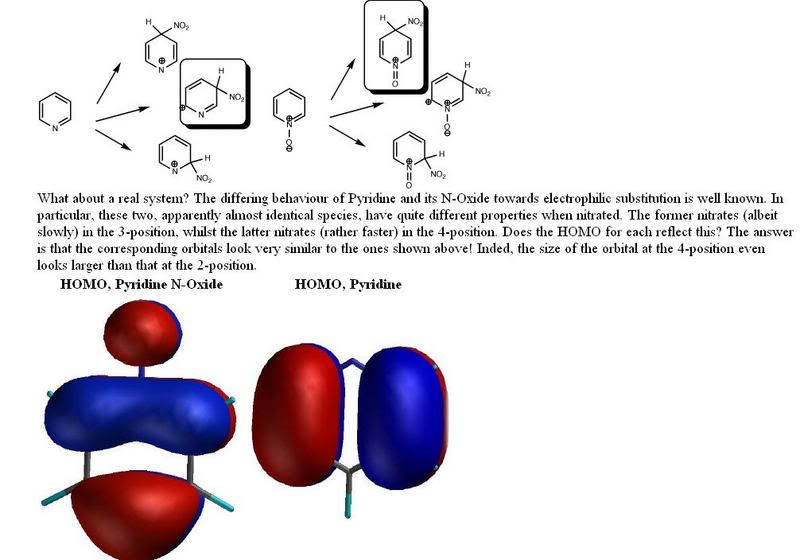
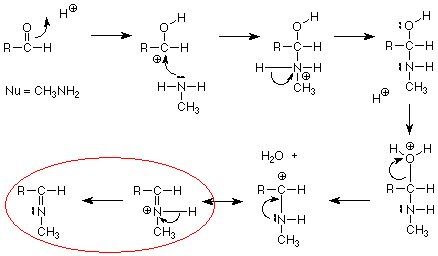

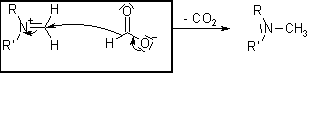

.gif "Matcuoi (95)") nên về sau cả nhóm thi nhau tìm phương pháp khác để chuyển amine nhất thành amine nhị đó ! mà cách methylation theo hoffmann mình vẫn chưa thử qua hỉ !
nên về sau cả nhóm thi nhau tìm phương pháp khác để chuyển amine nhất thành amine nhị đó ! mà cách methylation theo hoffmann mình vẫn chưa thử qua hỉ ! .gif "Matcuoi (14)")


 Linear Mode
Linear Mode

